# Kreatintransporterdefekt verstehen
Wichtiger Hinweis
Dieser Artikel behandelt ein Gesundheitsthema. Er dient weder der Selbstdiagnose noch wird die Diagnose eines Arztes ersetzt. Bitte beachten Sie hierzu unsere Allgemeinen und medizinischen Haftungsausschluss.
# Kreatin
# Allgemein
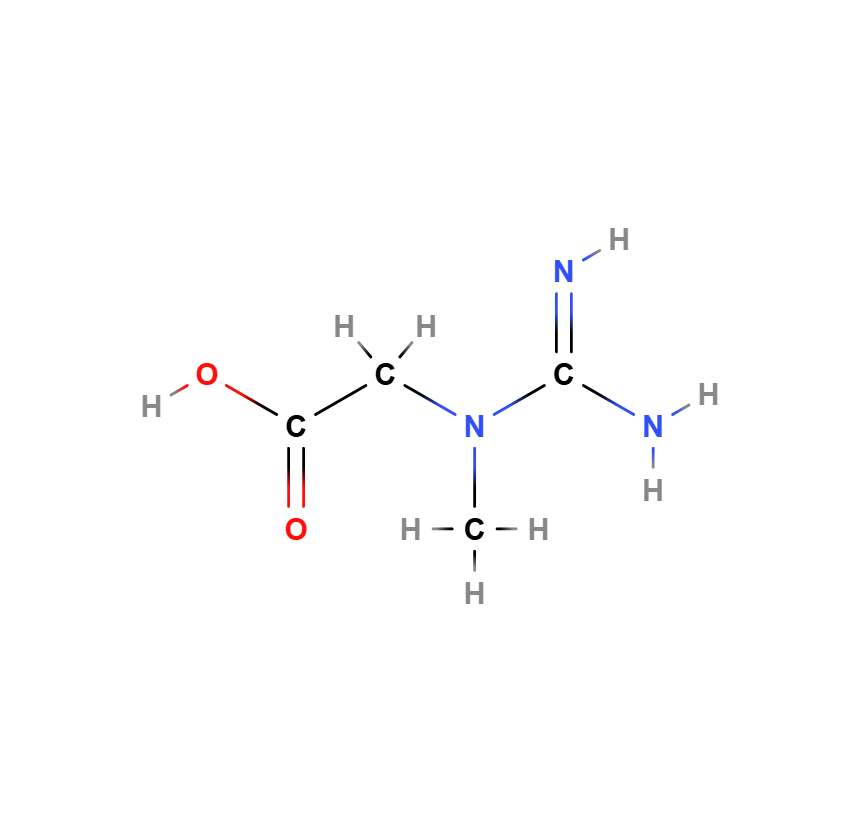
Kreatin (C₄H₉N₃O₂) ist eine stickstoffhaltige organische Säure, die aus den drei proteinogenen Aminosäuren Arginin, Glycin und Methionin gebildet wird. Haupteigenschaft für die Körperfunktion die die Versorgung der Körperzellen mit energiereichen Phosphanten. Es spielt eine essenzielle Rolle im Energiestoffwechsel der Zellen, insbesondere in Geweben mit hohem Energiebedarf wie Muskel- und Nervenzellen. Seine chemische Struktur enthält eine Guanidinogruppe und eine Carbonsäuregruppe, die für seine biologischen Eigenschaften wichtig sind.
Die Strukturformel zeigt:
- Eine Guanidinogruppe (-NH-C(=NH)-NH₂), die für die energetischen Eigenschaften von Kreatin entscheidend ist.
- Eine Carbonsäuregruppe (-COO⁻), die Wasserlöslichkeit und Aufnahme im Körper erleichtert.
# Physiologische Abhägigkeit von Transporterproteinen
Entscheidend ist, dass Kreatin durch seine chemische Struktur nicht in der Lage ist, die Doppellipidschicht der Zellmembran zu passieren. Kreatin ist ein hydrophiles Molekül mit polaren funktionellen Gruppen, insbesondere den Guanidinium- und Carbonsäuregruppen. Diese Gruppen interagieren stark mit Wasser und verhindern eine direkte Diffusion durch die lipophile Membran. Die Zellmembran besteht aus einer Phospholipid-Doppelschicht, deren hydrophobe Fettsäureketten als Barriere für polare und geladene Moleküle fungieren. Aufgrund dieser physikochemischen Eigenschaften kann Kreatin die Membran nicht ungehindert passieren. Um seine vielschichtigen physiologischen Aufgaben zu übernehmen, ist daher das Transporterprotein SLC6A8 notwendig, das Kreatin aktiv in die Zelle schleust.
 Abb.: Das Transporterprotein SLC6A8 ermöglicht die Passage vom extrazellulären Raum (z.B. Blut oder andere Zellen) in das Zytoplasma. Es ist essenziell, um das Kreatin-Phosphokreatin-System der Zellen aufrechtzuerhalten. Durch eine enzymkatalysierte Hydrolyse wird das Adenosintriphosphat-Molekül in Adenosindiphosphat und Phosphat gespalten (Creatin-Kinase). Quelle: Eigene Abbildung.
Abb.: Das Transporterprotein SLC6A8 ermöglicht die Passage vom extrazellulären Raum (z.B. Blut oder andere Zellen) in das Zytoplasma. Es ist essenziell, um das Kreatin-Phosphokreatin-System der Zellen aufrechtzuerhalten. Durch eine enzymkatalysierte Hydrolyse wird das Adenosintriphosphat-Molekül in Adenosindiphosphat und Phosphat gespalten (Creatin-Kinase). Quelle: Eigene Abbildung.
# Physiologische Bedeutung
# Allgemein
Kreatin spielt eine zentrale Rolle im Energiehaushalt der Zellen, insbesondere in Muskel- und Nervenzellen. Seine Hauptfunktion ist die Kurzzeitspeicherung und Bereitstellung von Energie durch das Phosphattransfersystem:
- Speicherung als Kreatinphosphat
- In den Zellen wird Kreatin durch das Enzym Kreatinkinase mit ATP (Adenosintriphosphat) zu Kreatinphosphat (CrP) phosphoryliert.
- Diese Verbindung dient als schnelle Energiereserve in Zellen mit hohem Energieverbrauch, insbesondere in Muskel- und Hirnzellen.
- Regeneration von ATP
- Bei hoher Belastung (z. B. intensiver Muskelarbeit) wird ATP verbraucht und in ADP (Adenosindiphosphat) umgewandelt.
- Kreatinphosphat gibt sein Phosphat an ADP ab, wodurch ATP regeneriert wird – ein schneller Energieschub für kurzzeitige, intensive Aktivitäten.
- Ausscheidung als Kreatinin
- Überschüssiges Kreatin wird nicht gespeichert, sondern zu Kreatinin abgebaut und über die Nieren mit dem Urin ausgeschieden.
- Der Kreatininspiegel im Blut gibt Hinweise auf die Nierenfunktion.
Die zellulären Kreatinreserven bei 70 kg Körpergewicht entsprechen durchschnittlich 120 g Kreatin. Die täglich abgebaute Menge an Kreatin beträgt ca. 1,7 bis 2 %. Mit einen Nettoausgleich von ca 2 Gramm Kreatin pro Tag werden die zellulären Kreatinreserven stabil gehalten (van de Kamp et al 2014 (opens new window)).
# Zelluläre Energiebereitstellung

Abb.: Anteile der verschiedenen zellulären Energiegewinnungsprozesse in Abhängigkeit von der Belastungsdauer (in Sekunden). Der orange Bereich zeigt den möglichen Energieverlust an, der ohne das vom Kreatintransporter abhängige Phospokreatin bestehen könnte. Eigene Abbildung.
1. ATP-Zerfall (orange Linie):
- Dieser Prozess stellt sofort verfügbare Energie z.B. bei der Muskelbewegung bereit, hält aber nur für sehr kurze Zeit (wenige Sekunden) bis das Adenosintriphosphat der Zelle aufgebraucht ist.
2. Kreatinphosphat-Zerfall (grüne Linie):
- Der sogenannte KP-Zerfall wird wwährend des ATP-Zerfalls aktiviert und liefert Energie für ca. 10 bis zu 30 Sekunden. Anschließend nimmt sein Anteil stark ab, bleibt aber aktiv.
- Dieser schnell verfügbare Energiegewinnungsprozess ist bei Kreatinmangelsydromen nicht oder in geringem Maße vorhanden.
3. Anaerobe Energiegewinnung (blaue Linie):
- Anaerobe Prozesse kommen verstärkt zum Einsatz, wenn sich die KP-Speicher reduzieren (ab Sekunde 10).
- Es handelt sich um die anaerobe Glykolyse, die ohne Sauerstoff abläuft und Laktat produziert.
4. Aerobe Energiegewinnung (rote Linie):
- Dieser Prozess wird mit zunehmender Belastungsdauer immer wichtiger.
- Er dominiert bei längerer Belastung (ab ca. 60 Sekunden), da er effizient und nachhaltig ist, aber mehr Zeit benötigt, um Energie bereitzustellen.
# Organische Wirkweise
# Bedeutung des Kreatin-Transporters für wesentliche Organe
Organische Funktionen, die der Kreatin-Transporter unterstützt, lassen sich wie folgt zusammenfassen (nach Farr et al 2020 (opens new window)):
- Skelettmuskulatur: Im muskulären Engeriestoffwechsel ist Keratin unter anderem als Teil des Kreatin-Phosphokreatin-Systems als Energiezwischenspeicher im flüssigen Zellplasma (zytosolischer Puffer) durch die Regeneration von ATP bei der Muskelkontraktion beteiligt (Wallimann et al 2011 (opens new window), Béard & Braissant 2010 (opens new window), Wyss & Kaddurah-Daouk 2000 (opens new window))
- Zentrales Nervensystem: Der Kreatin-Transporter übernimmt wesentliche Funktionen für Hirnzellen, wodurch höhere kognitive Funktionen ermöglicht werden. Das angereicherte Kreatin ermöglicht die ATP-Regeneration und den Transport von energiereichem Phosphat (ähnlich Skelettmuskulatur). Außerdem wird vermutet, dass Kreatin als Neurotransmitter fungiert, in Neuronen synthetisiert und in Abhängigkeit vom Aktionspotenzial freigesetzt werden kann (Béard & Braissant 2010 (opens new window)), da sich herausstellte, dass der Kreatin-Transporter an den Synapsen-Endköpfchen exprimiert wird. Möglicherweise, um das im synaptischen Spalt als Neurotransmitter freigesetzte und ungebundene Kreatin zu resorbieren bzw. zu recyceln (Hanna-El-Daher & Braissant 2016 (opens new window))
- Herzmuskulatur: Versorgung der Herzmuskulatur mit Kreatin.
- Embryonale Entwicklung: Kreatin-Transporter in der Plazenta nehmen Kreatin zur embyofetalen Entwicklung auf.
- Gastrointestinaltrakt: Versorgung der Darmepithelien mit Kreatin.
- Immunregulator: Kreatin hat regulierende Wirkung der Leukozyten und stabilisiert die Plasmamembranen der Erythrozyten.
- Nieren: Kreatin-Transporter resorbieren Kreatin aus dem Primär-Urin.
- Retina: Aufnahme von Kreatin in der Retina.
- Haut: Kreatin wirkt bei der Wundheilung mit und unterstützt den Hautschutz bei UV-Belastung
 Abb.: Anteile der verschiedenen zellulären Energiegewinnungsprozesse in Abhängigkeit von der Belastungsdauer (in Sekunden). Der orange Bereich zeigt den möglichen Energieverlust an, der ohne das vom Kreatintransporter abhängige Phospokreatin bestehen könnte. Eigene Abbildung nach (Farr et al 2020 (opens new window)).
Abb.: Anteile der verschiedenen zellulären Energiegewinnungsprozesse in Abhängigkeit von der Belastungsdauer (in Sekunden). Der orange Bereich zeigt den möglichen Energieverlust an, der ohne das vom Kreatintransporter abhängige Phospokreatin bestehen könnte. Eigene Abbildung nach (Farr et al 2020 (opens new window)).
# Quantisierung der Expression des Kreatin-Transporters in verschiedenen Gewebearten
Ein Hinweis auf die Bedeutung des Kreatintransporters für verschiedene Organe liefert das NIH Genotype-Tissue Expression (GTEx). Auch wenn seine Häufung in einer Gewebeart nicht unbedingt eine Aussage über die Funktionstychtigkeit des Organs aussagt, gibt sie jedoch Hinweise auf die Bedeuuntg des Transporters für das Gewebe.
 Abb.: Das Diagramm zeigt die Genexpression des SLC6A8-Gens in verschiedenen Geweben basierend auf Daten des GTEx (Genotype-Tissue Expression) Projekts an. Es handelt sich um ein Boxplot-Diagramm, das die Expression in Transcripts Per Million (TPM) für 54 verschiedene Gewebetypen darstellt (log10 Scalierung beachten!). Die Genexpression ist anhand der Median-Häufigkeit nach Gewebe sortiert. Genutzes Tool: GTEx Analysis Release V10.
Abb.: Das Diagramm zeigt die Genexpression des SLC6A8-Gens in verschiedenen Geweben basierend auf Daten des GTEx (Genotype-Tissue Expression) Projekts an. Es handelt sich um ein Boxplot-Diagramm, das die Expression in Transcripts Per Million (TPM) für 54 verschiedene Gewebetypen darstellt (log10 Scalierung beachten!). Die Genexpression ist anhand der Median-Häufigkeit nach Gewebe sortiert. Genutzes Tool: GTEx Analysis Release V10.
Die höchste Medianexpression von SLC6A8 über alle Stichproben tritt im Dickdarm, sigmoider Abschnitt auf (88.78 TPM). Auch in anderen Geweben des Verdauungstrakts gibt es eine vergleichsweise hohe Expression. In den meisten anderen Geweben ist die Expression niedriger oder sehr variabel (abhängig vom Individuum). Gelb markiete Bereiche im Diagramm sind Expressionen unterschiedlicher Regionen des Hirns. Das Cerebellum (Kleinhirn) zeigt eine etwas höhere Expression als andere Gehirnregionen. In der Großhirnrinde (Cortex) und dem Hippocampus gibt es eine breite Streuung, was auf variierende Expression zwischen Individuen hinweisen kann.
Näheres zu den Hirnregionen:
# Besondere Bedeutung von Kreatin für das Zentrale Nevensystem
ToDo
Die Bearbeitung der Inhalte ist noch in Arbeit
- Graphik der Synapsen
- Graphik der Bluthirnschranke
- Erläuterung
# Synthese und Exprimierung des Kreatintransporters
Das SLC6A8-Gen bildet den Natrium- und Chlorid-abhängigen Kreatintransporter. Es ist auf dem X-Chromosom, genauer in Region Xq28, lokalisiert. Es besteht aus 13 Exons, die die genetische Information für die Synthese des Kreatintransporters enthalten. Das daraus resultierende Protein besteht aus 635 Aminosäuren und hat eine molekulare Masse von ungefähr 70.523 Kilodalton (kD).
Die gesamte DNA-Sequenz des Gens umfasst etwa 8,4 Kilobasen (kb), während die mRNA, die als Bauplan für das Protein dient, eine Länge von ungefähr 3,9 kb aufweist.
Abb.: Die Abbildung ist mit GenCards und Alphafold entstanden und verbildlicht die vermutliche Faltung des Proteins. Blaue Signaturen sind hoch wahrscheinliche Strukturprognosen, während rötliche Bereiche Unsicherheiten in der tatsächlichen Faltung aufweisen.
# Synthese und Exprimierung

Abb.: Schematische Darstellung der Synthese von SLC6A8 und Exprimierung als Kreatintransporter an der Lipiddoppelschicht. Eigene Abbildung unter Nutzung von Bildausschnitten aus (Ulloa-Aguirre et al 2018 (opens new window))
Beschreibung Synthese und Exprimierung auf Grundlage diverser Veröffentlichungen von (Ulloa-Aguirre & Tao 2018 (opens new window)):
1. Gen-Transkription (Zellkern)
- Das Gen SLC6A8, das für das Transporterprotein kodiert, befindet sich in der DNA innerhalb des Zellkerns.
- Transkriptionsfaktoren binden an den Promotorbereich des Gens und leiten die Transkription ein.
- Die RNA-Polymerase transkribiert die DNA in Boten-RNA (mRNA) und für die Weiterbearbeitung und Tranapsort in Vesikeln im Zytoplasma modifiziert.
2a. mRNA-Export und -Übersetzung (Zytoplasma und Rough ER)
- Die verarbeitete mRNA verlässt den Zellkern durch die Kernporen und gelangt ins Zytoplasma.
- Ribosomen binden an die mRNA und beginnen mit der Translation.
- Der Transporter bindet das Ribosom an das raue endoplasmatische Retikulum (RER).
2b. Bei fehlerhafter Abschrift: Zersetzung und Recycling
- Falsch gefaltete oder falsch zusammengesetzte Molekülketten werden im ER zurückgehalten und den ansässigen Chaperonen ausgesetzt.
- Es erfolgt der Versuch die Faltung zu korrigieren und das Transporterprotein zu stabilisieren um den Export aus dem endoplasmatischen Retikulum zu ermöglichen.
- Wenn die korrekte Faltung fehlschlägt, wird das fehlgefaltete Protein in das Zytoplasma verschoben, wo es proteosomal (durch andere Proteine) abgebaut wird.
3. Proteinfaltung und posttranslationale Modifikationen (ER)
- Im RER wird das neu synthetisierte Polypeptid mit Hilfe von Chaperonproteinen in seine vorgesehene dreidimensionale Form gefaltet.
- Es finden posttranslationale Modifikationen statt, wie z. B. die Glykosylierung (Anbringung von Kohlenhydratgruppen) und die Bildung von Disulfidbindungen.
4. Protein-Transport und -Sortierung (Golgi-Apparat)
- Das Transporterprotein wird in Vesikeln vom ER zum Golgi-Apparat transportiert.
- Im Golgi können weitere Modifikationen wie Glykosylierung und Lipidanlagerung das Protein verfeinern.
- Der Golgi sortiert und verpackt den Transporter in Vesikel, die ihn an seinen endgültigen Bestimmungsort an der Zellwand bringen.
5. Vesikeltransport zur Plasmamembran
- Die Vesikel, die den Transporter enthalten, bewegen sich über das Zytoskelett (unter Beteiligung von Motorproteinen wie Kinesin oder Dynein) zur Plasmamembran.
- Das Vesikel verschmilzt mit der Plasmamembran und baut das Transporterprotein in die Lipiddoppelschicht (Zellwand) ein.
6. Funktionelle Expression an der Zelloberfläche
- Sobald der Transporter in die Plasmamembran eingebettet ist, ist er korrekt ausgerichtet und funktionsfähig.
- Er beginnt mit dem Transport von Kreatin durch die Zellmembran.
Regulierung und Recycling
- Das Transporterprotein kann durch Phosphorylierung, allosterische Modulation oder Wechselwirkungen mit anderen Proteinen reguliert werden.
- Bei Bedarf kann es durch Endozytose internalisiert und in Lysosomen abgebaut oder wieder in die Membran zurückgeführt werden.
# Mutation des Kreatintransporters
Ein Kreatintransporter-Defekt, der zu einer Kreatin-Transporter-Defizienz (CTD) führt, ist in der Regel auf Punktmutationen im SLC6A8-Gen zurückzuführen. Diese Mutationen können verschiedene Formen annehmen:
Nonsense-Mutation
Eine Nonsense-Mutation führt zu einem vorzeitigen Abbruch der Synthese des Kreatintransporters. Dies geschieht, wenn in der DNA fälschlicherweise ein Stopp-Codon eingefügt wird. Während der Translation durch die mRNA wird die Proteinsynthese dadurch frühzeitig beendet.
Bei CTD hat dies zur Folge, dass ein fehlerhaftes oder unvollständiges Transportprotein entsteht, welches zellintern durch das Qualitätskontrollsystem der Zelle abgebaut wird (siehe vorheriges Kapitel Punkt 2b und 6). Da es nicht korrekt in die Zellmembran eingebaut werden kann, bleibt die Transportfunktion aus.
Deletion/Insertion (Leserastermutation)
Hierbei wird entweder eine Base aus der DNA entfernt (Deletion) oder eine zusätzliche Base eingefügt (Insertion). Diese Veränderungen können das Leseraster des Gens verschieben, was häufig zur Produktion eines fehlerhaften oder funktionslosen Proteins führt.
- In vielen Fällen bricht die Synthese des Kreatintransporters vorzeitig ab (siehe vorheriges Kapitel Punkt 2b und 6).
- Alternativ kann der Transporter trotz Mutation gebildet und in die Zellmembran eingebaut werden, weist jedoch aufgrund von Fehlfaltung strukturelle Defekte auf, die seine Transportfunktion erheblich beeinträchtigen oder vollständig verhindern.
Missense-Mutation
Bei einer Missense-Mutation wird durch den Austausch einer einzelnen Base in der DNA ein verändertes Kodon erzeugt, welches für eine andere Aminosäure codiert. Dies kann zu strukturellen und funktionellen Änderungen des Proteins führen.
- Manche Missense-Mutationen beeinträchtigen die Transporterfunktion nur teilweise, sodass eine Restaktivität erhalten bleibt.
- Andere hingegen verursachen gravierende Fehlfunktionen oder führen dazu, dass das Protein instabil wird und vorzeitig abgebaut wird.
Missense-Mutationen können in ihrer Wirkung zwischen harmlos und schwerwiegend variieren, abhängig davon, welche Aminosäure ersetzt wird und welche Funktion die betroffene Region im Protein hat.

Abb.: Schematische Darstellung eines an der Lipiddoppelschicht exprimierten Transporters mit der Visualisierung einiger Genmutationen (und die Wirkung des synthetischen Chaperon 4-Phenyl-Butyric-Acid.). Eigene Abbildung.
# Therapieansätze
# Orale Gabe von Kreatinmonohydrat, L-Arginin und Glycin
In einer Studie wird 10 von 28 Patienten, die im Zuge einer hoch dosierten oralen Gabe von Kreatinmonohydrat zusammen mit L-Arginin und Glycin erhalten haben, eine klinische Verbesserung des Zustandes, z. B. durch eine Abnahme der Anfälle und Verbesserung der muskulären Symptome, beschrieben (Dunbar et al 2014 (opens new window), Valayannopoulos et al 2012 (opens new window)). Jedoch wird der oralen Gabe in diversen Untersuchungen eine Ineffektivität bzw. Unwirksamkeit dieser Behandlungsmöglichkeit im Hinblick auf eine Steigerung des Kreatingehalts im Gehirn bescheinigt (Jaggumantri et al 2015 (opens new window), Valayannopoulos et al 2012 (opens new window),van de Kamp et al 2011 (opens new window), Fons et al 2008 (opens new window), Bizzi et al 2002 (opens new window)). Es ist zu berücksichtigen, dass eine kombinierte Gabe von L-Arginin und Glycin bei einigen Patienten Hyperhomocysteinämie hervorgerufen hat (Villar et al 2011 (opens new window)). Hohe L-Arginin-Dosen über einem längeren Zeitraum setzen Patienten durch eine erhöhte Stickoxid-Synthese einem potenziellen Toxizitätsrisiko aus (Valayannopoulos et al 2012 (opens new window)).
Bei einer heterozygoten Patientin mit starker Epilepsie bewirkte die Gabe von Kreatin in Kombination mit Arginin und Glycin eine vollständige Auflösung der Anfälle (Mercimek-Mahmutoglu et al 2010 (opens new window)).
Als Hauptursache für den ausbleibenden Anstieg des zerebralen Kreatins wird diskutiert, dass im Zuge des Kreatin-Transportes über die Blut-Hirn-Schranke (BHS), bis zu drei bzw. fünf Membranen zu passieren sind (mit bzw. ohne Sternzellen) (Fernandes-Pires & Braissant 2022 (opens new window)), die ohne funktionales Transporter-Protein nicht oder in zu geringer Menge überwindbar sind: Von den mikrokapillaren Endothelzellen an der BHS zu den umgebenden Astrozyten und zu den Gehirnzellen im ZNS-Parenchym (z. B. Neuronen und Oligodendrozyten).
# Entwicklung von lipophilen Kreatin-Analoga
Kreatin-Analoga sind der Grundsubstanz Kreatin sehr ähnlich und weisen daher vergleichbare chemische Eigenschaften auf. Von einigen lipophilen Kreatin-Derivaten wird angenommen, dass sie eine gute Permeabilität durch die Doppellipidschicht der Zellmembran aufweisen. Dadurch könnte es möglich sein, auch ohne den Kreatin-Transporter den Kreatingehalt an den organischen Wirkungsorten zu erhöhen. Die Wirkweise ist in obiger Abbildung unter Punkt 2 modellhaft zu sehen. Lipophile Kreatin-Analoga könnten sich somit als ein vielversprechender Arzneimittelkandidat erweisen (Trotier-Faurion et al 2013 (opens new window)).
# Verbindung des Kreatins mit Salzen
Durch Konjugation von Kreatin mit Salzen kann eine Aufnahme von Kreatin über andere Transporter erfolgen. In obiger Abbildung ist dies durch Punkt 3 dargestellt.
# Pharmakologische Chaperone
Bei pharmakologischen Chaperonen handelt es sich um kleine Moleküle, die die Fehlfaltung des mutierten Proteins verhindern/korrigieren, seine Translokation an die richtige zelluläre Lokalisation ermöglichen und dafür sorgen können, dass es seine Aktivität wiedererlangt. Sie können sich daher als therapeutischer Ansatz für Patienten mit Fehlfaltungsmutationen von SLC6A8-Varianten eigenen, in dem sie bei der Faltung während der Genese des Transporters frühzeitig im endoplasmatischen Retikulum wirken oder nach Lokalisation eines fehlgefalteten Kreatin-Transporters an der Zellmembran Faltungskorrekturen vornehmen (siehe obige Abbildung Punkt 4). Für einige Mutationen, die zu Fehlfaltungen des Transporters führten, wurde mit 4-Phenyl-Butyric-Acid (4-PBA) und Hitzeschockproteine experimentiert (Farr et al 2020 (opens new window)), um die Fehlfaltungen zu korrigieren. Auch wenn die genauen Wirkmechanismen von 4-PBA noch weitgehend unbekannt sind, beweisen Untersuchungen mit dem synthetischen Chaperon 4-PBA grundsätzlich, dass Fehlfaltungen am SLC6A8-Protein behoben werden können. Vorausgesetzt, es handelt sich um eine Mutationsvariante, die auf Proteinfaltungsfehler im endoplasmatischen Retikulum zurückzuführen ist (El-Kasaby et al 2019 (opens new window)). Ursächlich ist hierfür, dass 4-PBA im endoplasmatischen Retikulum wirkt und die dort durchgeführte Faltung des Transporterproteins beeinflussen kann.
# Gentherapie
Aktuell sind mehrere Ansätze in Arbeit, die auf adeno-assoziierte Viren (AAV) basieren. Sie werden bereits als viraler Vektor in der Gentherapie verwendet und haben sich als einer der am häufigsten und sichersten Vektoren erwiesen (Fernandes-Pires & Braissant 2022 (opens new window)). In Hinsicht auf den Kreatin-Transporter-Defekt sind die besonderen Vorteile der AAV, dass sie sich leicht manipulieren lassen, um ein Transgen zur Genübertragung in die Zellen/Zellkerne einzuführen und durch sie Neuronen mit der gewünschten Gensequenz zu infizieren. Damit könnte ermöglicht werden, dass funktionsfähige Transporter-Proteine in den Hirnzellen etabliert werden können, wodurch die Patienten ausreichende Mengen an zellulären Kreatin aufbauen könnten.
# Behandlung von Patienten
Einschlägige Fachliteratur
Bitte nutzen Sie die medizinische Fachliteratur, beachten Sie ggü. älteren Studien Neuerungen bei der oralen Gabe von Kreatinmonohydrat mit Sublementen. Ggf. liegen aktuellere Verfahrensweisen vor. Wenden Sie sich an medizinische Fachexperten oder nehmen Sie für weitere Informationen und für Vermittlungsgesuche Kontakt mit uns auf.